Genomes of viruses show a high mutation rate at the nucleotide level, which can be exploited for molecular typing purposes. By sequencing a specific variable region of the viral genome, one can investigate whether or not selected isolates of a certain species are genetically related or not.
Sequence-based typing of viruses is a quick and portable method used in outbreak settings, e.g. to investigate nosocomial infections in hospitals and other health care facilities. BIONUMERICS offers a rich databasing and analysis platform, where sequence data sets can be compared, clustered and analyzed in conjunction with any other phenotypic, genotypic, epidemiologic and geographic data. Exchange data between users and laboratories through BIONUMERICS’ database sharing tools and set up molecular surveillance networks (e.g. CaliciNet).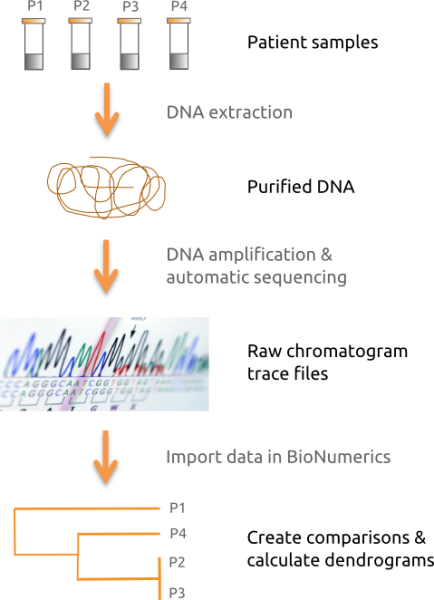
Sequence-based typing in BIONUMERICS
BIONUMERICS allows batch import and assembly of raw chromatogram files using the batch sequence assembly plugin. Reference sequences can be imported from online repositories or from FASTA and GenBank file formats. Sequences can be manually annotated in the sequence viewer window. Perform multiple alignments using well-established algorithms, such as Needleman-Wunsch and Wilbur-Lipman or our own proprietary algorithm.
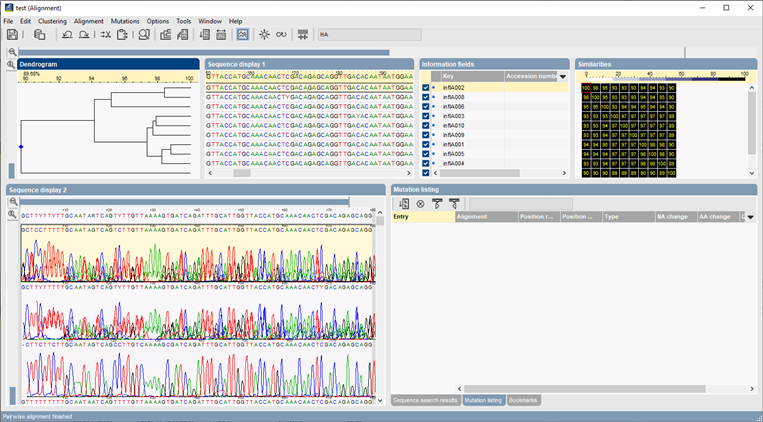
Create comparisons and calculate similarity or distance matrices and dendrograms for any selected group of sequences using a large number of statistical coefficients and clustering methods (UPGMA, Ward, Neighbor Joining, etc.). Alternatively, create Minimum Spanning Trees for plotting epidemic divergence against factors such as geographical distribution, date of sampling, serotypes, etc.