Multi Locus VNTR Analysis (MLVA) is a molecular typing method to subtype microbial isolates based upon the Variable copy Numbers of Tandem Repeats (VNTR). A VNTR typically exhibits a large range of copy numbers, even among highly related bacterial strains. For a selected set of tandem repeats, copy number analysis reveals insights about the relationships at a micro-evolutionary level.
")
In practice, VNTR loci are selected that are sufficiently and complementary discriminatory for the organisms studied, and conserved primers are designed outside the tandem repeat for each VNTR. Thus, the size in base pairs of each PCR-amplicon is the sum of the size of the tandem repeat plus the offsets at both ends.
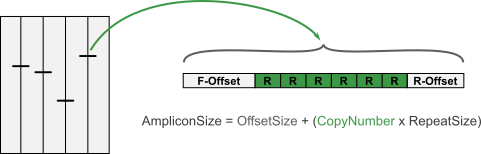
Knowing the repeat size, the copy number can easily be calculated as

For economy reasons, several VNTRs are sometimes pooled, i.e. they are marked with the same dye and loaded as a mixture in the same column of a capillary sequencer. A condition is that the mixed VNTR PCR products have size ranges that do not overlap. E.g., using 4 dyes and 2 non-overlapping VNTRs, 6 VNTRs can be determined per capillary run (one dye contains a reference marker set for size calculation).
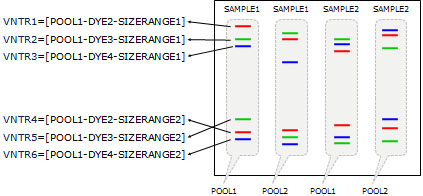
Multilocus VNTR analysis in BIONUMERICS
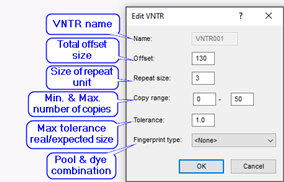
The BIONUMERICS software offers a fully automated workflow for multi-locus VNTR analysis, starting from raw capillary sequencer chromatogram files or preprocessed peak tables (Applied Biosystems and Beckman). The MLVA setup has to be entered initially in the database. This involves entering the pooling strategy: a pool is a mix of VNTR amplification products loaded together in the same capillary. This includes the different dyes used and optionally, the compatible VNTRs with non-overlapping size ranges. Thus, each VNTR is defined by a pool, a dye and (optionally) a size range. The size range is defined by the repeat length, the offset and the copy range. As such, the software knows exactly within which size range it should look for a specific VNTR. Note that the copy range is only essential in case different VNTRs are pooled with the same dye.
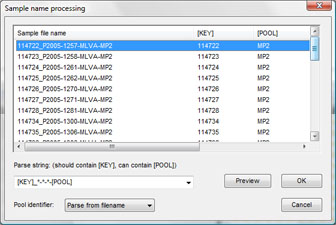 In case of raw chromatogram files (AB, Beckman), the software can automatically parse pool, dye and strain information from the file names, using a parsing string defined by the user.
In case of raw chromatogram files (AB, Beckman), the software can automatically parse pool, dye and strain information from the file names, using a parsing string defined by the user.
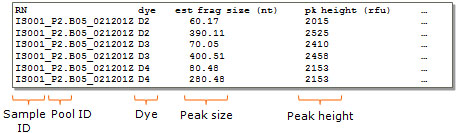
In case of GeneMapper or Beckman peak table files, this information is automatically parsed from the tab-delimited peak table (see example below).
Robust and reliable approach, independent of instrument type
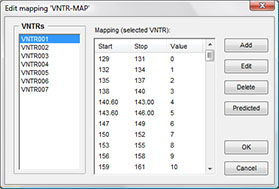 Due to differences in instruments, dyes, and capillary columns, measured VNTR amplicon sizes often differ more or less from theoretical sizes based upon copy numbers. Therefore, a tolerance can be entered in bp. Obviously, the tolerance should always be less than RepeatSize/2. In case of small repeat sizes, calculated copy numbers may be systematically different when compared between different instrument types. To solve this problem, BIONUMERICS offers the possibility to create VNTR maps, i.e. a mapping from observed size to real size per VNTR and per copy number. Such VNTR ensure compatibility between different instruments, protocols, dyes, columns etc.
Due to differences in instruments, dyes, and capillary columns, measured VNTR amplicon sizes often differ more or less from theoretical sizes based upon copy numbers. Therefore, a tolerance can be entered in bp. Obviously, the tolerance should always be less than RepeatSize/2. In case of small repeat sizes, calculated copy numbers may be systematically different when compared between different instrument types. To solve this problem, BIONUMERICS offers the possibility to create VNTR maps, i.e. a mapping from observed size to real size per VNTR and per copy number. Such VNTR ensure compatibility between different instruments, protocols, dyes, columns etc.
Fully automated copy number analysis
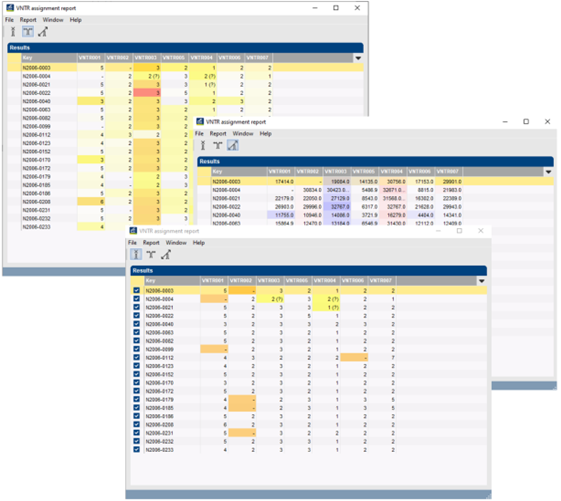
Once the settings for VNTRs and parsing have been entered, the software can automatically process thousands of MLVA runs, thereby creating reports listing unresolved VNTRs, multiple peaks found, and any other problems. Reports can display deviation from expected value as green to red (below, upper image), left/right deviation (blue and red, resp. (center image) or only errors and warnings (bottom image).
A myriad of analysis tools
The resulting VNTR information is stored in integer-type character sets where each VNTR represents one character. VNTR data can be analyzed as categorical characters (each different copy number is a different allele) or as quantitative characters. In the latter case, the larger the difference between copy numbers, the less related the organisms are considered. Population modelling networks can be calculated using the finest and most comprehensive cluster analysis application available today, applying micro-evolutionary criteria as priority rules and displaying branch significance support indication. The Minimum Spanning Tree algorithm applied on VNTR data in BIONUMERICS has proven to be invaluable for epidemiological study and population genetics of bacterial populations.
Improved MLVA in BIONUMERICS
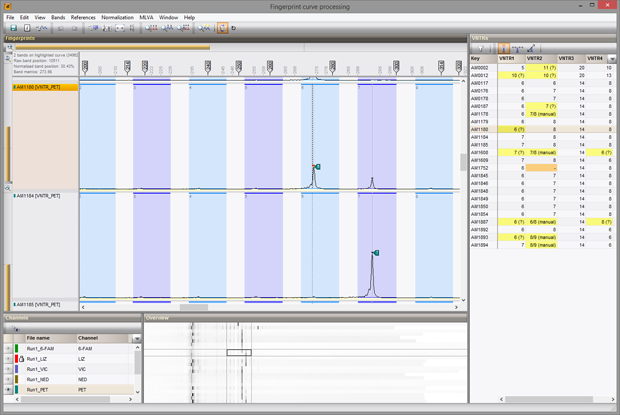
In close collaboration with our customers, we have completely redesigned the MLVA plugin to integrate seamlessly with the Curve processing window, which was introduced in version 7.0 as a dedicated electropherogram processing environment. The result is a more flexible setup and an improved workflow, leading to faster and more accurate VNTR copy number determination.
Easy setup of one or multiple MLVA schemas per database
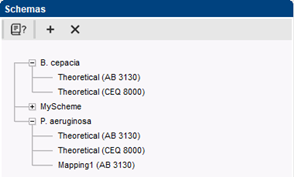 In contrast to the previous implementation, more than one MLVA schema can be used in the same database. This is useful for example when a database combines isolates from several species and each species has its own MLVA schema.
In contrast to the previous implementation, more than one MLVA schema can be used in the same database. This is useful for example when a database combines isolates from several species and each species has its own MLVA schema.
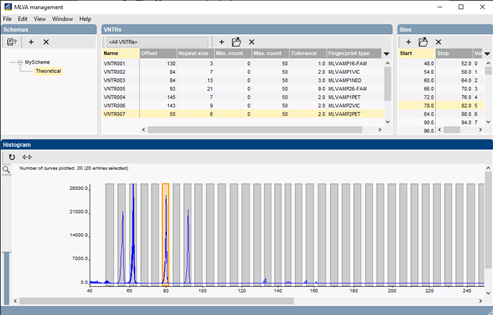
The new MLVA management window provides direct access to all relevant MLVA settings. Each MLVA schema has its theoretical VNTR bin prediction, based on repeat lengths, tolerances and offsets for each VNTR. Additionally, a MLVA schema can have one or more custom mappings to accommodate for deviations in fragment size determination. Custom mappings can be tweaked by drag-and-drop bin positioning based on the normalized curves. For labs working with more than one capillary sequencer, multiple machine types can optionally be defined.
Complete MLVA schemas can easily be exchanged via import/export of XML files. This ensures consistency between different databases and different researchers working with the same organism.
MLVA-based typing
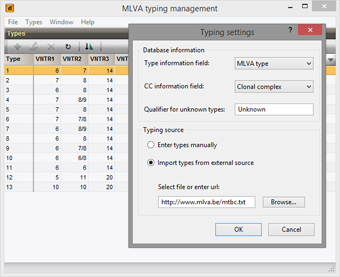 Similar as for MLST, assigning types can greatly facilitate communication e.g. in epidemiologic studies. MLVA types and clonal complexes can be assigned based on the complete set or any subset of VNTRs.
Similar as for MLST, assigning types can greatly facilitate communication e.g. in epidemiologic studies. MLVA types and clonal complexes can be assigned based on the complete set or any subset of VNTRs.
To facilitate the use of a uniform and stable nomenclature, MLVA types can be imported and synchronized from external files or URLs.
Dealing with double VNTR assignments
In clinical samples or with certain organisms, the presence of double VNTR alleles is observed. The new MLVA plugin allows you to make such double VNTR assignments.
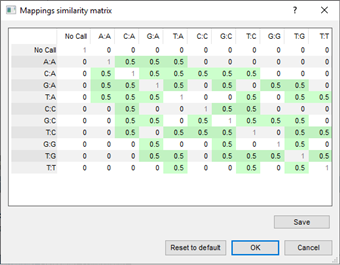
In comparisons and similarity calculations, double assignments are dealt with via character mapping similarity matrices, which can be customized by the user.
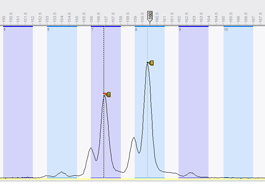
Enhanced import and processing of electropherograms
In BIONUMERICS, the import of electropherograms is integrated in the general import functionality, making it both easier and more flexible.
Small but noticeable enhancements in the Curve processing window were introduced, from which MLVA users no doubt will benefit. These include different sets of band search parameters for size markers and samples separately and an easier way to select/unselect peaks.